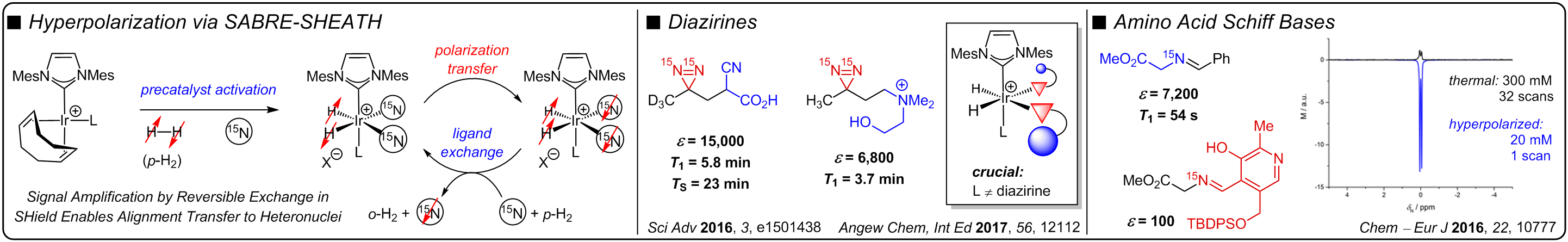
UMPOLUNG AMINE SYNTHESIS
A traditional approach for the stereoselective synthesis of chiral amines via C–C bond formation employs the addition of nucleophiles to electrophilic imines. Although a number of elegant reactions have been developed that utilize this strategy, the polarity of the disconnection inherently limits the chemical space accessible, including the synthesis of vicinal amino alcohols and diamines, among others. Many of these motifs serve as important precursors to medicinally important scaffolds. Our lab is interested in developing reverse polarity methods for constructing chiral amines that utilize α-amino anion synthons, facilitating catalyst and/or reagent controlled stereoselective synthesis of a range of challenging amine-containing motifs. Specifically, we have discovered that 2-azadienes may be used as enamine umpolung reagents, enabling the enantio- and diastereoselective synthesis of sterically congested 1,2-amino tertiary alcohols and 1,2-diamines. We have also utilized 2-azaallyl anions as imine umpolung reagents for the diastereoselective and stereospecific synthesis of stereochemically dense 1,3-amino alcohols and diamines.
CATALYTIC ENANTIOSELECTIVE DIENE HYDROFUNCTIONALIZATION
The enantioselective assembly of compounds bearing allylic stereogenic centers is an important objective in organic chemistry, both because of their utility in downstream synthesis and because of their prevalence in natural products and bioactive molecules. The in situ generation of allyl–transition metal complexes offers an attractive means of furnishing these moieties either through the electrophilic trapping of a nucleophilic crotylating agent or nucleophilic addition to a metal–π-allyl (Tsuji-Trost allylic substitution). Rarely do these reactions deliver compounds with 1,2-disubstituted olefins. In our lab, we seek to develop catalytic enantioselective intermolecular diene hydrofunctionalizations, transformations that are atom economic, proceed under mild reaction conditions, and afford myriad small molecule building blocks comprised of internal olefins and allylic stereogenic centers. We have discovered that aliphatic amines and activated carbon pronucleophiles may be added to acyclic dienes with high degrees of enantio- and site-selectivity when promoted by Pd catalysts bearing electron-deficient PHOX ligands.

HYPERPOLARIZATION VIA SABRE-SHEATH
The sensitivity of NMR/MRI methods is limited, due in part to the small thermal fractional magnetization of nuclei, resulting in a severe detection limit for abundant nuclei (e.g., 1H), a situation which is exacerbated for rarer isotopes, such as 13C and 15N. Hyperpolarization methods may overcome these issues by increasing the fractional magnetization of nuclei, and SABRE-SHEATH, discovered by the Warren lab at Duke, shows great promise as a cost-effective and operationally simple way of achieving this. In this process, spin order is catalytically transferred from para-hydrogen gas to a molecule of interest by the transient binding of both to an Ir complex, allowing polarization buildup on the substrate. Working with the Warren (chemical physics) and Wang (chemical biology/chemical synthesis) labs at Duke, as well as the Theis lab at NC State, our research group is interested in the design and development of new Ir complexes for this process as well as enhancing our understanding of the fundamental organometallic chemistry. Initial studies in the hyperpolarization of diazirines and amino acid derivatives show large signal enhancements (ε) over the thermally-acquired signal for long lifetimes in several cases.